Theorietag 2
Auf 9 Seiten gibt es den folgenden Theorietext auch zum Download.
Der Text hier auf der Homepage ist älter und etwas
anders. Er stimmt aber hinreichend mit der Druckversion überein:
Drei
organische Verbindungstypen mit der sauerstoffhaltigen funktionellen Gruppe
am Ende:
Alkanole > Alkanale > Alkansäuren am Beispiel Ethanol > Ethanal >
Ethansäure:
Strukturformel, Alltagsgegenwart von Ethanol / Isopropylalkohol / Formaldehyd / Essig
Theoretische Chemie: Formal wird da Kohlenwasserstoff schrittweise oxidiert.
Oxidation wurde jahrhundertelang verstanden als Sauerstoffaufnahme. Modern
ist es „Abgabe von Außenelektronen an einen Bindungspartner“. Oxidation ist
immer damit gekoppelt, dass etwas reduziert wird. Bei unseren O-haltigen
organischen Verbindungen muss also einem anderen Stoff Sauerstoff entzogen
worden sein. Modern ist diese „Reduktion“ eines Stoffes „Aufnahme von
Außenelektronen von einem Bindungspartner“.
Das passierte im Experiment bei der Verbrennungsgruppe: Paraffin wurde umgeben
von schwarzem Kupferoxid und hoch erhitzt. Es entstanden kurzkettige Alkanole
und Aldehyde. Das Reaktionsprodukt wurde vom Lehrer aufgehoben und kann am
Praxistag 2 betrachtet werden.
Exemplarisch (das ist kein Klausurthema, außer die Grupe wünscht es) werden an die C-Atome von Ethanol > Ethanal > Ethansäure, und bei Bedarf an weitere Strukturformeln (Di-Ethyl-Ether, Aceton und Ethansäure-Ethylester) die Oxidationszahlen geschrieben.
Drei Organische Verbindungen mit der sauerstoffhaltigen funktionellen Gruppe in der Mitte:
Ether - Ketone - Ester am Beispiel Di-Ethyl-Ether, Aceton und Ethansäure-Ethylester.
Alltagsgegenwart: Ether als historisches Betäubungsmittel, in der Forschung als
Extraktionsmittel. Aceton als Nagellackentferner. Ester als Aromastoffe und
Bestandteil von Klebstoffen.
Bezug zum Praktikumstag: Aceton löst auch spielend Styropor,
zeigte die „Lösungsgruppe“. Ester lassen sich minutenschnell herstellen mit
Hilfe des Katalysators Schwefelsäure, zeigte die „Estergruppe“.
Exkurs: Herstellen von Estern als Aromastoffe für Lebensmittel - da darf keine Schwefelsäure rein - durch stundenlanges Kreisen im Rückflusskühler. Recht nett dazu die „Synthese eines Duftstoffes“ unter dem Motto „Frauen schnuppern in Naturwissenschaften“ http://www.chemie.unibas.ch/~nachwuchs/chemie/modul_5.html .
Theorie: Was ist allgemein ein Katalysator? Enzyme als Katalysatoren - im Menschen spezialisiert auf Zusammenarbeit bei 37 Grad Körpertemperatur.
Exkurs: Was ist Extraktion? Verständnis für Teebeutel, Kaffee, Ölzugabe beim Salat, Verzehren einer Karotte zugleich mit einem Butterbrot.
Einzeichnen der funktionellen Gruppen „Hydroxylgruppe“ - „Aldehydgruppe“ - „Säuregruppe“ - „Ethergruppe“ - „Ketogruppe“ - „Estergruppe“ in allen sechs Beispielsubstanzen.
Herleitung von Ethergruppe und Estergruppe als Kondensationsreaktionen: Alkohol wird „verethert“ unter Wasseraustritt - je ein Molekül Alkansäure und Alkanol wird „verestert“ unter Wasseraustritt. Der Katalysator Schwefelsäure als „Wasserräuber“ bei der Veresterung.
Herleitung der Ketogruppe als finales Oxidationsprodukt eines „sekundären Alkanols“ - danach geht nur noch ein Zerreißen des Moleküls. Zeichnung eines „tertiären Alkanols“ - das kann nicht mal ein Aledyd werden.
Mehrwertige Alkanole als Strukturformel: Glykol und Glycerin. Exemplarische Benennung (kein Klausurthema) Propan-2-ol (Trivialname Isopropanol), Ethan-1,2-diol (Trivialname Glykol), Propan-1,2,3-triol (Trivialname Glycerin). Die Zahl vor der Endung -ol gibt die Position der funktionellen Gruppe(n) an.
- Pause -
Vergleich der sechs Stoffgruppen mit Sauerstoff dabei, die im Blockkurs vorgestellt wurden, nach drei Kriterien:
1. Die jeweils gegebene homologe Reihe anhand einer gleichförmig wachsenden C-Kettenlänge. Es gibt auch immer eine gemeinsame Summenformel.
2. Ist
eine homologe Reihe gegeben, verändern sich die physikalischen Eigenschaften
in stetiger Weise.
Hierzu ein Vergleich der Siedepunkte von Alkanen und Alkanolen im gleichen
Diagramm. Das erheblich höhere Sieden der Alkanole, „nur“ weil sie ein
Sauerstoff-Atom an Bord haben, fällt auf.
3. Das vergleichbare chemische Reaktionsverhalten von Stoffen, die der
gleichen homologen Reihe angehören.
In unserem Praktikum hatten wir hierzu
- die im Prinzip stets gleiche Technik der Veresterung, egal welches Alkanol man
auf welche Alkansäure treffen lässt.
- die mit jedem Alkan gleich machbare Oxidation mit schwarzem Kupferoxid zu
Alkanol und Alkanal
- das für jedes Alkanal nutzbare Fehling-Reagenz.
Exkurs: Was ist ein Reagenz? Ein Stoff, der einen andern Stoff selektiv, auch in störender Umgebung, nachweist. Die Gruppe „Alkanole - Alkanale“ hat hierfür ein Beispiel: Mit Borax brennt Methanol in einer anderen Farbe als das im Bau und in der Chemie benachbarte Ethanol.
Nach
Beispielen für 3. erst gehen wir zurück zu Kriterium 2. Damit war die
„Lösungsgruppe“ befasst. Die Hydroxylgruppe ist „hydrophil“. Sie
ermöglicht ein Bindungsverhalten zwischen Alkanolen, wie es bei Wasser gegeben
ist. Das Gegenteil davon ist „hydrophob“, wasserfürchtend. Alle
Kohlenwasserstoffe sind hydrophob.
Benzin verteilt sich auf einer Pfütze zu einer einmolekularen Schicht - was die
bekannten Regenbogenfarben von Benzin auf Wasser bewirkt. Eine Abschätzung der
Zahl der Benzin-Moleküle, die sich in einem Kubikzentimenter Benzin befinden,
ist damit möglich (Ölfleckversuch, Loschmid-Zahl).
Moleküle
können aneinander haften durch folgende drei Kräfte:
- Wasserstoffbrückenbindungen „WBBs“. Die entstehen zwischen H-Atomen an
unsymmetrisch gebautem Molekül mit polarer Atombindung: 25 KJ/Mol
- Dipolkräfte. Die enstehen zwischen unsymmetrisch gebauten Molekülen mit
polarer Atombindung: 10 KJ/Mol
- Van-der-Waals-Kräfte. Die sind im Prinzip bei allen Molekülen gegeben.
Sie sind bei kleinen Molekülen aber sehr schwach - bei Wasser unter 1 KJ/Mol.
Sie wachsen mit der Masse des Moleküls und mit seinem Oberflächenkontakt.
Also C5H12 Pentan hat schon 3 KJ/Mol VdW-Kräfte, und C20H42
Paraffin hat dann 50 KJ/Mol VdW-Kräfte.
2,2-Dimethyl-Propan hat aufgrund seiner kugeligen Außenfläche nur 2 KJ/Mol
VdW-Kräfte im Verglich zum gleich schweren n-Pentan mit seinen 3 KJ/Mol.
Moleküle mischen sich so gut, wie sich die Kräfte gleichen, die sie zusammenhalten („similia similibus solvuntur“). Wasser und Glycerin mischen sich z.B. beliebig. Kohlenwasserstoffe mit ihren reinen VdW-Kräften hingegen verhalten sich abweisend gegenüberWasser, das fast nur durch WBBs und Dipolkräfte seinen Zusammenhalt hat.
Manche Stoffe liegen zwischen beiden Welten - das Lösungsmittel Aceton z.B.. Es hat erhebliche Dipolkräfte, aber keine WBBs. Es mischt sich mit Benzin, aber auch mit Wasser. Bei Alkohol ist bekannt, dass er in feuchter Umgebung nicht lange rein bleibt: Er nimmt 3 Prozent Wasser auf. Dieses Gemisch hat günstigere zwischenmolekulare Kräfte als reiner Alkohol. Mischt man 500 ml Alkohol mit 500 ml Wasser, erhält man 980 ml Gemisch, und nicht 1000 ml ...
Alkanole haben also durch ihre Hydroxylgruppe mit ihren WBBs und Dipolkräften einen wesentlich größeren zwischenmolekularen Zusammenhalt als Alkane mit ihren Vdw-Kräften. Die bei gleicher Zahl der C-Atome sehr verschiedenen Schmelz- und Siedepunkte beider homologer Reihen nähern sich erst bei langkettigen Verbindungen einander an: Da wird die Wirkung der einen Hydroxylgruppe immer belangloser.
Die „Lösungsgruppe“ demonstriert uns Mischbares und Mischungsfeindliches. Darüber hinaus zeigt sie am Bespiel der Reaktion von Natrium in Wasser und in Ethanol, inwieweit die Hydroylgruppe des Ethanols zu chemisch vergleichbarem Verhalten - in diesem Fall mit Wasser - führt.
An dieser Stelle lässt sich auch der Versuch der Arbeitsgruppe „Herstellen von Ethin“ besprechen. Im Prinzip können wir uns einige chemische Gleichungen anschauen:
Die Herkunft des CaC2 aus dem Hochofenprozess: CaCO3 + 3 C + FeO --> CaC2 + Fe + 2 CO2 (CaCO3 wird im Hochofen zum Erniedrigen der Schmelztemperatur von FeO = Eisenerz zugegeben)
Die Reaktion im Rundkolben: CaC2 + 2 H2O --> Ca(OH)2 + C2H2
Die Verbrennung des Ethingases an Luft: 2 C2H2 + 3 O2 --> 2 CO2 + 2 H2O + 2 C (das sind die Rußflocken)
All diese Gleichungen werden nicht in der Klausur gefragt, Sie stehen nur mal hier.
Zwei Fragen ergeben sich aus dem Beitrag dieser Gruppe:
1. Wieso explodiert ein Gas-Luft-Gemisch, während sich ein reines Gas, das in Luft hinausströmt, ohne Explosion entzünden lässt?
2. Wie kann aus so wenig Feststoff CaC2 so viel Gas C2H2 entstehen?
Das beantworten wir bei Zeit und Interesse. Dann ist erst mal
- Pause -
Mit der bisherigen Theorie am zweiten Theorie-Tag haben wir die Tür aufgestoßen zum schnellen Verständnis von zwei komplexen biologisch-organischen Stoffgruppen:
1.
Fette sind Ester zwischen je einem Glycerinmolekül und drei Fettsäuren.
Fettsäuren sind langkettige Alkansäuren, Beispiel Stearinsäure.
Zeichnung der Strukturformel eines Fettes. Erläuterung: Gesättigte contra
ungesättigte Fettsäuren. Ungesättigte Fettsäuren haben ein bis drei
Doppelbindungen und sind essentiell.
Exkurs: „Essentielle“ Stoffe kann der Mensch - können zumeist Tiere allgemein nicht selbst im Körper herstellen. Sie müssen sie mit der Nahrung aufnehmen. Essentiell sind für uns einige Aminosäuren, zwei ungesättigte Fettsäuren, Mineralstoffe, Spurenelemente und die bekanntesten, die Vitamine.
Kleiner Gag: Ratten können Vitamin C selbst bilden. Es ist nicht essentiell für sie, aber für den Menschen: Wenn wegen Vitamin-C-Mangel auf Schiffen im Mittelalter Skorbut ausbrach, hatten die Ratten kein Problem...
Erläuterung: Wie kommt der Körper darauf, so ein vertracktes Molekül wie „Fett“ so viel zu verwenden?
- Es ist nicht wasserlöslich und lässt sich deshalb gut im bekanntlich sehr wasserhaltigen Körper lagern. Fett ist unsere Vorratsenergie.
- Es hat
einen doppelt so hohen „Brennwert“ wie Proteine oder Kohlenhydrate. Also
Fette sind unsere Kalorienbomber. Das ist für fast alle Lebewesen außer für sich
zu wenig bewegende zu viel auf Essangebote reagierende Menschen sinnvoll.
(Nachfrage: Ist allen der Brennwert in KJ ein Begriff aus dem Alltag?)
- Es lässt sich chemisch aus dem Ende der Glykolyse heraus, vor Beginn des Zitratzyklus, über die „aktivierte Essigsäure“ aufbauen - und rückwärts auf diesem Wege auch wieder in den Stoffwechsel einschleusen. Wegen des Aufbaus aus aktivierter Essigsäure, einem C-2-Molekül, sind alle im Menschen vorkommenden Fettsäuren gradzahlig: 12, 14, 16, 18, 20, 22 als C-Kettenlängen.
Kurz oder tief, je nach Wunsch: Ausflug in die Glykolyse und den Zitratzyklus. Das sind Themen der Biologie - „Zellatmung“ und „Mitochondrien als Kraftwerke“. Es wird aber in der mündlichen Biologie-Prüfung nur sehr oberflächlich gefragt, da einige Biologie-Lehrer einen Bogen darum machen.
Uns ist im Praktikum die Glykolyse bei der Gärung begegnet. Da zerlegen die Enzyme der Hefe nämlich Zucker. Die Hefe kann sich daraus Energie in Form von ATP holen. Wenn Sauerstoff fehlt, kommt die Zuckerzerlegung bei einem C2-Molekül zum Erliegen. Da also könnte die Hefe, wie wir jetzt wissen, auch Fett aufbauen. Typischerweise aber stellt sie als Endprodukt C2H5OH her - also Alkohol.
2. Zucker sind zumeist ringförmig gebaute mehrwertige Alkanole mit einer einzelnen Aldehyd- oder Ketogruppe. Da der Ringschluss einer C-Kette am besten mit 6 beteiligten Molekülen funktioniert, enthalten die typischen Zucker 5 oder 6 C-Atome.
Exkurs: Ringschluss bei Kohlenwasserstoffen - das Benzolmolekül, die Aromaten.
Beispiele für Zucker:
Ribose bei RNA, Desoxyribose bei DNA (zwei fast gleiche Fünfer-Zucker),
Glucose und Fructose (Trauben- und Fruchtzucker, zwei Sechser-Zucker),
sowie dann Zweierzucker = Disaccharide (Saccharose besteht aus einem Molekül Glucose und einem Molekül Fructose, verbunden über eine Etherbrücke (die heißt nun „glycosidische Bindung“).
Der
typische Zucker passt in die Formel CnH2nOn - Glucose als Beispiel: C6H12O6.
Das sieht von außen so aus, als klebte an jedem C-Atom ein H2O-Wassermolekül.
Irrtrümlich wurde die Stoffgruppe 1844 daher als „Kohlenhydrate“ bezeichnet, was
man aber bis heute nicht geändert hat.
Süß schmecken uns Mono- und Disaccharide.
Mit glykosidischer Bindung können dann aber auch Polysaccharide aufgebaut werden:
- das
schnell auf- und abbaubare, vielfach verzweigte Glykogen in Tieren
- der Lagerstoff „Stärke“, wenig verzweigt, in Pflanzen.
Mögliche
Exkurse:
- Wie stellt man aus Stärke Malzzucker her?
- Warum beginnt Brot beim Dauerkauen süß zu schmecken?
- Historische Nutzung von Stärke als Kleister.
Auf Wunsch: Der Gesamtbau eines Nukleotids: Im Zentrum die Ribose, zum Verknüpfen die Phosphorsäuregruppe - sie kann einen Ester durch Kondensationsreaktion mit dem Nachbarmolekül herstellen - und als kennzeichnendes Molekül eine der vier Basen A, T, C, G bei DNA bzw. A, U, C, G bei RNA.
Nur zum Anschauen, nicht zum Lernen: Einfachringbau der Pyrimidinbasen Thymin und Cytosin, Doppelringbau der Purinbasen Adenin und Guanin. Zwei Wasserstoffbrücken zwischen A und T, drei Wasserstoffbrücken zwischen C und G.
Krönender möglicher Abschluss: ein chemisch korrektes ganzes DNA-Nukleotid.
- Pause -
Der erste Theorietag kam fast ganz mit Kohlenwasserstoffen aus. Der zweite Theorietag gelangte bisher sehr weit, indem er nur mal Sauerstoff als dritten Bindungspartner einführte.
Zum Verständnis von Aminosäuren und ihrer Verkettung zu Proteinen brauchen wir nun als Bindungspartner noch Stickstoff. Der steckt in der Aminogruppe und ist eine Base.
Biologische Aminosäuren haben einen „konstanten Bereich“, der immer gleich gebaut ist und es ermöglicht, dass Aminosäuren durch die Kondensationsreaktion zwischen einer Aminogruppe und einer Säuregruppe lange Ketten bilden. Dabei ragt der jeweile „besondere Bereich“ einer Aminosäure aus der Kette heraus und bewirkt das typische physikalische und chemische Verhalten der AS-Kette.
Alle Lebewesen bauen ihre Proteine durch die gleichen 20 AS auf. Es gibt hydrophile und hydrophobe, polare (Dipolkräfte!) und unpolare, saure und basische „besondere Bereiche“.
Zeichnung von Alanin mit hydrophobem Bereich - eine Methylgruppe, Serin mit hydrophilem Bereich - eine Hydroxylgruppe.
Exkurs: Säure, Base, PH-Wert, Indikator.
Zeichnung der Entstehung einer Peptidbindung.
Kurzes Erwähnen von Primär- Sekundär- und Tertiärstruktur eines Proteins. Das ist Thema des Standart-Biologie-Unterrichtes und nicht des Chemie-Blockkurses.
Im Prinzip gibt es auch anders gebaute AS als diejenigen in der Biologie. Mit ihnen kann man Kunststoffe herstellen. Der in der Biologie übliche Begriff „Peptidbindung“ beschreibt denselben Bindungstyp wie die Amidbindung dieser Polyamide. Bei heutigen Polyamiden werden allerdings zwei Komponenten - zum einen ein Molekül mit zwei Carbonsäuregruppen, und zum andern ein Molekül mit zwei Aminogruppen, zur Kondensationsreaktion miteinander verrührt.
Na, war das eine trickreiche Überleitung vom Protein zum Kunststoff?
Seit etwa 1950 stecken wir in der Kunststoff-Revolution (Nobelpreis 1953 an Hermann Staudinger). Das Leben mit Holz, Gips, Metall, Beton, Leinen und Wolle war hölzern dagegen. Im Einsatz sind Kunststoffe seit etwa 1900, indem man sie zufällig erfand: Bakelit ist der bekannteste. Den Bau und einige Herstellungsweisen von Kunststoffen richtig formuliert hatte Staudinger von 1920 bis 1932. Er wurde aber von den Nazis lahmgelegt: „Hitlers Wissenschaftler hatten keinen Draht zum Kunststoff“
26 Millionen Tonnen Kunststoff in Deutschland pro Jahr verteilen sich in folgende Bereich der Wirtschaft:
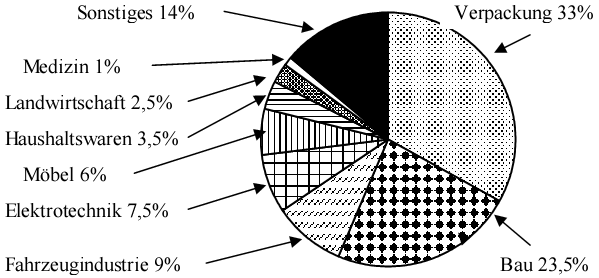
Allen Kunststoffen liegt das gleiche Prinzip zugrunde: Kleine Moleküle verknüpfen sich zu Makromolekülen. Aus Monomer wird Polymer.
Im typischen Fall wird die Verarbeitung der Monomere zu Polymeren nur von wenigen großen spezialisierten Fabriken durchgeführt (z.B. BASF, Evonik). Denn es ist ein oft giftiger High-Tech-Prozess. Sodann erhalten zahlreiche Einzel-Hersteller das schon auspolymerisierte „Kunststoff-Granulat“, behandeln es mit Additiven weiter und verformen es. Das klappt bei Thermoplasten.
Preiswerte Kunststoffe haben ein Hitzeproblem. Die Welt des Kunststoffs liegt unter 100 Grad Celsius. Es gibt keine Automotoren aus Kunststoff.
Spezielle Kunststoffe haben ein Giftproblem. Von PVC (tötet bei Brand) über formaldehyhaltige Kunststoffe (allergiefördernd) bis zu Teflon (als Krebsauslöser verdächtig) zeigen sich bei vielen Kunststoffen Nebenwirkungen.
Umgekehrt ist die Vielfalt der Aufgaben, die sich mit Kunststoffen lösen lassen, gegenüber andern Materialien überwältigend, ob in Technik, Medizin, Design, Spielzeug.
Kunststoffe sind so preiswert, dass sie lange für alle Wegwerfartikel eingesetzt wurden, und erst heute ein wenig Umdenken hin zu abbaubaren Verpackungen einsetzt. Der typische Kunststoff ist so wenig abbaubar wie ein Stein. In Flüssen gibt es zwischen Steinen mittlerweile abgeschabte Kunststoffpartikel.
Nur Kunststoffe, die sich nicht verformen lassen (Duroplaste), werden am Ort der Endproduktion aus Momomeren hergestellt: Kunststoffböden, Ausschäumen von Innenteilen mit Hartschaum, Klebstoffe. Soweit Duroplaste sich sägen und polieren lassen - Plexiglas, Resopal - werden sie dann aber auch schon fertig polymerisiert geliefert.
Zur Herstellung von Duroplasten werden an Ort und Stelle zwei Komponenten miteinander vermengt, die zuvor getrennt gehalten werden müssen: Monomer 1 und 2. Manchmal ist als drittes noch ein „Starter“ der Reaktion dabei. Diese Komponenten polymerisieren dann unumkehrbar.
Durch Wahl passender Monomere erhält man eine gewünschte chemische Eigenschaft - z.B. „linear“ für Fäden und Tüten, „verzweigt“ für räumliche Gebilde und Beschichtungen.
Drei chemische Reaktionsypen gibt es bei der Herstellung von Kunststoffen:
1. Die Polykondensation. Zwei Monomere treffen aufeinander. Beide Momomere haben mindestens je zwei funktionelle Gruppen. An den Bindungsstellen zum benachbaerten Monomer wird ein kleines Molekül abgespalten, z.B. Wasser.
Polyester entstehen so: Ein Diol oder Polyol trifft auf eine Dicarbonsäure oder Polycarbonsäure. Ein Diol ist ein Molekül mit zwei Hydroxylgruppen. Glykol hatten wir da kurz kennengelernt. Poly-ol und Di... Polycarbonsäure lassen sich entsprechend definieren.
Reaktionsprinzip bei der Bildung eines Polyesters:
HO - (CnH2n) - OH (das Diol) + HOOC - (CnH2n) - COOH (die Dicarbonsäure) --->
HO -
(CnH2n) - CO - O - (CnH2n) - CO - O - (CnH2n) - CO - O - (CnH2n) - COOH
(der Polyester mit links und rechts einem offenen Ende zur Weiterreaktion)
2. Die Polyaddition. Hier reagieren Monomere mit passenden funktionellen Gruppen so miteinander, das keine Neben- und Spaltprodukte freiwerden (wie es bei der Polykondensation der Fall ist).
Diese „Monomere mit passenden funktionellen Gruppen“ übersteigen aber das, was wir mit unserem Sauerstoff und Stickstoff bisher zu sehen bekamen. Die in Matratzen elastischen, in Hartschäumen stabilen Polyurethane nutzen z.B. die funktionelle Gruppe „Isocyanat“ und reagieren mit den Hydroxylgruppen eines zweiten Momomers:
O=C=N-(CH2)6-N=C=O + HO-(CH2)4-OH ----> -(CH2)6-NH-CO-O-(CH2)4-
Hinter dem Reaktionspfeil ist eine durch Addition entstandene Bindung zu sehen. Das H der Hydroxylgruppe ist jetzt am N, und alle Atome sind noch da, kein Stoff spaltet sich ab. Nach beiden Seiten läuft das so weiter.
3. die Polymerisation. Sie arbeitet im typischen Fall mit gleichen Monomeren, die Doppelbindungen haben. Da ist die „radikalische Polymerisation“ machbar, bei der die Doppelbindungen mit je einer ihrer zwei Bindungen sich mit dem Nachbar-Monomer verbinden. Eine radikalische Reaktion haben wir schon bei der Bromierung von Hexen kennengelernt: Da öffnet sich die Doppelbindung und greift sich das Brom-Molekül.
Über radikalische Polymerisation aus Ethen-Monomeren stellt man z.B. „Polyethen“ her, den meistgenutzten Kunststoff überhaupt:
Im Lauf der Reaktion: Einsatz einer radikalischen Startersubstanz
C=C + C=C + C=C + C=C ---------------------------------> -C-C-C-C-C-C-C-C- usw.
Kunststoffe werden dreigeteilt nach ihrem Verhalten bei Zug und Stoß:
Duroplaste können splittern. Im leicht erkennbaren Fall haben sie
Werkeigenschaften ähnlich wie Holz.
Sie haben ausschließlich festen Aggregatszustand.
Bei Erhitzen zersetzen sie sich.
Mit Lösungsmitteln quellen sie auf.
Thermoplaste sind teils elastisch, also sie nehmen ihre vorherige Form nach Zug oder Stoß wieder ein, teils sind sie verformbar. Oft haben sie eine Erweichungstemperatur irgendwo zwischen 80 und 160 Grad Celsius: Darunter verhalten sie sich elastisch, darüber kann man sie verformen.
Thermoplaste werden bei Erhitzen schrittweise weich, aber nie flüssig. Bei zu
hohem Erhitzen zersetzen sie sich.
In Lösungsmitteln kann man sie gelegentlich auflösen, häufiger nur deformieren.
Insbesondere Thermoplaste werden zentral hergestellt, als Granulat transportiert
und in verteilten Fabriken weiter verarbeitet.
Alle denkbaren Verarbeitungsmethoden kommen zum Einsatz mit Pressformen,
Tiefziehen mit Vakuum, Walzen zu Folien und Spinnen von Fäden.
Elastomere: Das ist eine trickreiche kleine Sondergruppe der Kunststoffe, die Gummis. Sie sind sehr dehnbar, und nach dem Ende von Zug oder Stoß nehmen sie ihre vorherige Form wieder ein. Man kann Elastomere überdehnen: Dann reißen sie, oder sie verlieren ihre hochelastische Eigenschaft.
Klassisches Gummi wird aus Rohkautschuk durch Vulkanisation hergestellt: Der Saft des Kautschukbaumes wird mit Schwefel verrührt. Der Schwefel stellt Polymere her mit „biegbaren Atombindungen“ - Bindungsarme, die nicht starr ihren Bindungswinkel brauchen, sondern schwenkbar sind - bei Druck oder Zug biegen sie sich passend. In einem Polymer-Netzwerk können zuvor rundliche Strukturen so enorm länglich werden: „Aus Kugel wird Röhre“.
Die elastischen Eigenschaften von Gummi können durch Einlagern von Ruß beeinflusst werden. Durch Rußeinlagerung sind Autoreifen schwarz - wie übrigens auch Vinyl-Schallplatten (Polyvinyl ist allerdings ein Thermoplast).
Klassische Gummis altern an Luft, weil mit den Jahren der chemisch
vergleichbare Sauerstoff (6. Hauptgruppe wie Schwefel) den Schwefel ersetzt. Wo
Gummi alterungsbeständig sein soll, behilft man sich mit Silikon-Gummi-Sorten.
Die sind aber teurer und nie so elastisch.
Wir deuten hier eine zweite, benachbarte Kunststoff-Chemie an, die auf
verketteten Silízium-Atomen statt Kohlenstoff-Atomen beruht - und beenden da das
Thema.
Farben
Vor vielen hundert Jahren waren bunte Dinge (Kleider, Oberflächen, Malfarben) zur Hälfte bunt durch „mineralische“ Stoffe - Salze und Metalle: Ultramarinblau, Chromgrün, Asbestweiß. Organische Farben stellten die andere Hälfte (Purpurrot, Indigoblau).
Heutzutage beruhen 99 Prozent des Bunten um uns auf organischer Chemie (was denn nicht? Das Eloxalverfahren braucht anorganische Chemie).
Damit ein Stoff bunt erscheint statt weiß oder schwarz, muss er Teile des Lichtes (das ist „weiß“) aufnehmen (= absorbieren), und andere Teile zurückwerfen, uns also vor Augen führen (= reflektieren).
„Licht“ ist der für unser Auge sichtbare Bereich des viel weiteren Spektrums der elektromagnetischen Wellen. Elektromagnetische Wellen sind teils kürzer als Licht. Da reichen sie von Gamma-Strahlung (10 -13 m) über Röntgen- ( 10 -10 m) bis zur UV ( 10 -7 m) - Strahlung.
Andererseits können elektromagnetische Wellen langwelliger sein als Licht: Das beginnt bei Infrarot (10 -5 m) und geht über die Radarstrahlung (1 cm, gilt auch für Mikrowelle) bis hin zu den Funkwellen UKW, KW, MW und LW (Wellenlänge 1 m bis 1 km).
Unser sichtbares Licht hat ein Spektrum vom kurzwelligen Blau (400 nm = 4 x 10-6 m) bis zum langwelligen Rot (680 nm).
Wenn ein Stoff rot, grün und gelb absorbiert, blau aber reflektiert, sieht er blau aus. Mischfarben wie Braun entstehen durch Pigmente, die mehrere kleine Abschnitte des Lichtspektrums reflektieren und Dazwischenliegendes absorbieren.
Wissenswert ist Chlorophyll: Das ist grün, weil es alle Farben außer Grün absorbiert, also für die chemische Reaktion „Photosynthese“ nutzen kann. Nur Grün wird reflektiert. Bestrahlt man eine grüne Pflanze mit grünem Licht, verkümmert sie.
Organische Stoffe haben einen typischen Trick, wie sie sich bunt machen: Die konjugierte Doppelbindung. Sie tritt in organischen Molekülen dann auf, wenn Einfach- und Doppelbindung sich abwechseln. Das haben wir bei „Butadien“ am ersten Theorietag schon mal kurz erwähnt, und unser theoretischer Rundlauf durch die organische Chemie schließt sich hier.